Sickle-cell disease (SCD), or sickle-cell anemia, is an autosomal recessive genetic blood disorder with overdominance characterized by red blood cells that assume an abnormal, rigid, and sickle shape. Sickling decreases the cells' flexibility and results in a risk of various complications, including shortened life expectancy. It is caused by a change in hemoglobin's primary structure .
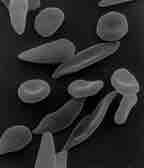
Sickle cell anemia is caused by a change in hemoglobin's primary structure
This scanning electron micrograph shows red blood cells. Some exhibit the normal donut-shaped morphology while others exhibit the sickle shape characteristic of those with sickle-cell trait or sickle-cell disease.
Disease Presentation
Sickle-cell disease, usually presenting in childhood, occurs more commonly in people (or their descendants) from parts of tropical and sub-tropical regions where malaria is or was common. One-third of all indigenous inhabitants of Sub-Saharan Africa carry the gene because, in areas where malaria is common, there is a fitness benefit in carrying only a single sickle-cell gene (sickle cell trait). Those with only one of the two alleles of the sickle-cell disease, while not totally resistant, are more tolerant to malarial infection and show less severe symptoms when infected. The prevalence of the disease in the United States is approximately 1 in 5,000, mostly affecting Americans of Sub-Saharan African descent, according to the National Institutes of Health. In the United States, about 1 out of 500 African-American children born will have sickle-cell anemia.
The term "sickle cell crisis" is used to describe several independent acute conditions occurring in patients with sickle cell disease. Sickle cell disease results in anemia and several types of crises. Most episodes of sickle cell crises last between five and seven days. The vaso-occlusive crisis is caused by sickle-shaped red blood cells that obstruct capillaries and restrict blood flow to an organ, resulting in ischaemia, pain, necrosis and often organ damage. The frequency, severity, and duration of these crises vary considerably. Because of its narrow vessels and function in clearing defective red blood cells, the spleen is frequently affected in splenic sequestration crisis. This autosplenectomy increases the risk of infection from encapsulated organisms. Preventative antibiotics and vaccinations are recommended for those with such asplenia. Aplastic crises are acute worsenings of the patient's baseline anemia producing pallor, tachycardia, and fatigue. This crisis is triggered by parvovirus B19, which directly affects erythropoiesis (production of red blood cells) by invading the red cell precursors and multiplying in them and destroying them. Parvovirus infection nearly completely prevents red blood cell production for two to three days. In normal individuals, this is of little consequence, but the shortened red cell life of sickle-cell patients results in an abrupt, life-threatening situation. Hemolytic crises are acute accelerated drops in hemoglobin level. The red blood cells break down at a faster rate. This is particularly common in patients with co-existent G6PD deficiency. Management is supportive, sometimes with blood transfusions.
Diagnosis and Prevention
Sickle cell can be diagnosed by a blood test. In HbSS, the full blood count reveals hemoglobin levels in the range of 6–8 g/dL with a high reticulocyte count (as the bone marrow compensates for the destruction of sickle cells by producing more red blood cells). In other forms of sickle-cell disease, Hb levels tend to be higher. Abnormal hemoglobin forms can also be detected on hemoglobin electrophoresis, a form of gel electrophoresis on which the various types of hemoglobin move at varying speeds.
People who are known carriers of the disease often undergo genetic counseling before they have a child. A test to see if an unborn child has the disease takes either a blood sample from the fetus or a sample of amniotic fluid. Since taking a blood sample from a fetus has greater risks, the latter test is usually used. Children born with sickle-cell disease will undergo close observation by the pediatrician and will require management by a hematologist to assure they remain healthy. These patients will take a 1 mg dose of folic acid daily for life. From birth to five years of age, they will also have to take penicillin daily due to the immature immune system that makes them more prone to early childhood illnesses.
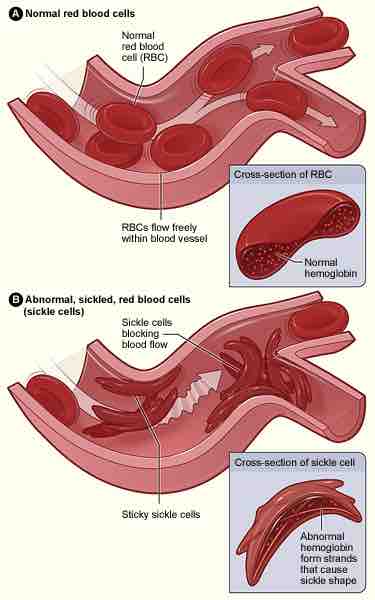
Sickle-cell disease
Figure A shows normal red blood cells flowing freely in a blood vessel. The inset image shows a cross-section of a normal red blood cell with normal hemoglobin. Figure B shows abnormal, sickled red blood cells blocking blood flow in a blood vessel. The inset image shows a cross-section of a sickle cell with abnormal (sickle) hemoglobin forming abnormal strands.