
Actinide
Background Information
SOS Children, an education charity, organised this selection. A quick link for child sponsorship is http://www.sponsor-a-child.org.uk/
| Actinides in the periodic table | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

The actinide or actinoid (IUPAC nomenclature) series encompasses the 15 metallic chemical elements with atomic numbers from 89 to 103, actinium through lawrencium.
The actinide series derives its name from the group 3 element actinium. All but one of the actinides are f-block elements, corresponding to the filling of the 5f electron shell; lawrencium, a d-block element, is also generally considered an actinide. In comparison with the lanthanides, also mostly f-block elements, the actinides show much more variable valence.
| 89 Ac |
90 Th |
91 Pa |
92 U |
93 Np |
94 Pu |
95 Am |
96 Cm |
97 Bk |
98 Cf |
99 Es |
100 Fm |
101 Md |
102 No |
103 Lr |
Of the actinides, primordial thorium and uranium occur naturally in substantial quantities and small amounts of persisting natural plutonium have also been identified. The radioactive decay of uranium produces transient amounts of actinium and protactinium, and atoms of neptunium, americium, curium, berkelium and californium are occasionally produced from transmutation reactions in uranium ores. The other actinides are purely synthetic elements. Nuclear weapons tests have released at least six actinides heavier than plutonium into the environment; analysis of debris from a 1952 hydrogen bomb explosion showed the presence of americium, curium, berkelium, californium, einsteinium and fermium.
All actinides are radioactive and release energy upon radioactive decay; naturally occurring uranium and thorium, and synthetically produced plutonium are the most abundant actinides on Earth. These are used in nuclear reactors and nuclear weapons. Uranium and thorium also have diverse current or historical uses, and americium is used in the ionization chambers of most modern smoke detectors.
In presentations of the periodic table, the lanthanides and the actinides are customarily shown as two additional rows below the main body of the table, with placeholders or else a selected single element of each series (either lanthanum or lutetium, and either actinium or lawrencium, respectively) shown in a single cell of the main table, between barium and hafnium, and radium and rutherfordium, respectively. This convention is entirely a matter of aesthetics and formatting practicality; a rarely used wide-formatted periodic table inserts the lanthanide and actinide series in their proper places, as parts of the table's sixth and seventh rows (periods).
Discovery, isolation and synthesis
| Element | Year | Method |
|---|---|---|
| Neptunium | 1940 | Bombarding 238U by neutrons |
| Plutonium | 1941 | Bombarding 238U by deuterons |
| Americium | 1944 | Bombarding 239Pu by neutrons |
| Curium | 1944 | Bombarding 239Pu by α-particles |
| Berkelium | 1949 | Bombarding 241Am by α-particles |
| Californium | 1950 | Bombarding 242Cm by α-particles |
| Einsteinium | 1952 | As a product of nuclear explosion |
| Fermium | 1952 | As a product of nuclear explosion |
| Mendelevium | 1955 | Bombarding 253Es by α-particles |
| Nobelium | 1965 | Bombarding 243Am by 15N or 238U with α-particles |
| Lawrencium | 1961–1971 | Bombarding 252Cf by 10B or 11B and of 243Am with 18O |
Like the lanthanides, the actinides form a family of elements with similar properties. Within the actinides, there are two overlapping groups: transuranium elements, which follow uranium in the periodic table—and transplutonium elements, which follow plutonium. Compared to the lanthanides, which (except for promethium) are found in nature in appreciable quantities, most actinides are rare. The most abundant, or easy to synthesize actinides are uranium and thorium, followed by plutonium, americium, actinium, protactinium and neptunium.
The existence of transuranium elements was suggested by Enrico Fermi based on his experiments in 1934. However, even though four actinides were known by that time, it was not yet understood that they formed a family similar to lanthanides. The prevailing view that dominated early research into transuranics was that they were regular elements in the 7th period, with thorium, protactinium and uranium corresponding to 6th-period hafnium, tantalum and tungsten, respectively. Synthesis of transuranics gradually undermined this point of view. By 1944 an observation that curium failed to exhibit oxidation states above 4 (whereas its supposed 6th period neighbour, platinum, can reach oxidation state of 7) prompted Glenn Seaborg to formulate a so-called "actinide hypothesis". Studies of known actinides and discoveries of further transuranic elements provided more data in support of this point of view, but the phrase "actinide hypothesis" (the implication being that "hypothesis" is something that has not been decisively proven) remained in active use by scientists through the late 1950s.
At present, there are two major methods of producing isotopes of transplutonium elements: irradiation of the lighter elements with either neutrons or accelerated charged particles. The first method is most important for applications, as only neutron irradiation using nuclear reactors allows the production of sizeable amounts of synthetic actinides; however, it is limited to relatively light elements. The advantage of the second method is that elements heavier than plutonium, as well as neutron-deficient isotopes, can be obtained, which are not formed during neutron irradiation.
In 1962–1966, there were attempts in the United States to produce transplutonium isotopes using a series of six underground nuclear explosions. Small samples of rock were extracted from the blast area immediately after the test to study the explosion products, but no isotopes with mass number greater than 257 could be detected, despite predictions that such isotopes would have relatively long half-lives of α-decay. This inobservation was attributed to spontaneous fission owing to the large speed of the products and to other decay channels, such as neutron emission and nuclear fission.
From actinium to neptunium

Uranium and thorium were the first actinides discovered. Uranium was identified in 1789 by the German chemist Martin Heinrich Klaproth in pitchblende ore. He named it after the planet Uranus, which had been discovered only eight years earlier. Klaproth was able to precipitate a yellow compound (likely sodium diuranate) by dissolving pitchblende in nitric acid and neutralizing the solution with sodium hydroxide. He then reduced the obtained yellow powder with charcoal, and extracted a black substance that he mistook for metal. Only 60 years later, the French scientist Eugène-Melchior Péligot identified it with uranium oxide. He also isolated the first sample of uranium metal by heating uranium tetrachloride with potassium. The atomic mass of uranium was then calculated as 120, but Dmitri Mendeleev in 1872 corrected it to 240 using his periodicity laws. This value was confirmed experimentally in 1882 by K. Zimmerman.
Thorium oxide was discovered by Friedrich Wöhler in the mineral, which was found in Norway (1827). Jöns Jacob Berzelius characterized this material in more detail by in 1828. By reduction of thorium tetrachloride with potassium, he isolated the metal and named it thorium after the Norse god of thunder and lightning Thor. The same isolation method was later used by Péligot for uranium.
Actinium was discovered in 1899 by André-Louis Debierne, an assistant of Marie Curie, in the pitchblende waste left after removal of radium and polonium. He described the substance (in 1899) as similar to titanium and (in 1900) as similar to thorium. The discovery of actinium by Debierne was however questioned in 1971 and 2000, arguing that Debierne's publications in 1904 contradicted his earlier work of 1899–1900. The name for word actinium comes from the Greek aktis, aktinos (ακτίς, ακτίνος), meaning beam or ray. This metal was discovered not by its own radiation but by the radiation of the daughter products. Owing to the close similarity of actinium and lanthanum and low abundance, pure actinium could only be produced in 1950. The term actinide was probably introduced by Victor Goldschmidt in 1937.
Protactinium was possibly isolated in 1900 by William Crookes. It was first identified in 1913, when Kasimir Fajans and Oswald Helmuth Göhring encountered the short-lived isotope 234mPa (half-life 1.17 minutes) during their studies of the 238U decay. They named the new element brevium (from Latin brevis meaning brief); the name was changed to protoactinium (from Greek πρῶτος + ἀκτίς meaning "first beam element") in 1918 when two groups of scientists, led by Otto Hahn and Lise Meitner of Germany and Frederick Soddy and John Cranston of Great Britain, independently discovered 231Pa. The name was shortened to Protactinium in 1949. This element was little characterized until 1960, when A. G. Maddock and co-workers in UK produced 130 grams of protactinium from 60 tonnes of waste left after extraction of uranium from its ore.
Neptunium (named for the planet Neptune, the next planet out from Uranus, after which uranium was named) was discovered by Edwin McMillan and Philip H. Abelson in 1940 in Berkeley, California. They produced the 239Np isotope (half-life 2.4 days) by bombarding uranium with slow neutrons. It was the first transuranium element produced synthetically.
Plutonium and above

Transuranium elements do not occur in sizeable quantities in nature and are commonly synthesized via nuclear reactions conducted with nuclear reactors. For example, under irradiation with reactor neutrons, uranium-238 partially converts to plutonium-239:
![\mathrm{ {}^{238}_{92}U + {}^{1}_{0}n\ \xrightarrow\ \ {}^{239}_{92}U\ \xrightarrow[23.5\ min]{\beta^-}\ {}^{239}_{93}Np\ \xrightarrow[2.3\ days]{\beta^-}\ {}^{239}_{94}Pu\ \xrightarrow[2.4\cdot 10^4\ years]{\alpha} }](../../images/1195/119579.png)
In this way, Enrico Fermi with collaborators, using the first nuclear reactor Chicago Pile-1, obtained significant amounts of plutonium-239, which were then used in nuclear weapons.
Actinides with the highest mass numbers are synthesized by bombarding uranium, plutonium, curium and californium with ions of nitrogen, oxygen, carbon, neon or boron in a particle accelerator. So, nobelium was produced by bombarding uranium-238 with neon-22 as
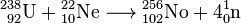 .
.
First isotopes of transplutonium elements, americium-241 and curium-242, were synthesized in 1944 by Glenn T. Seaborg, Ralph A. James and Albert Ghiorso. Curium-242 was obtained by bombarding plutonium-239 with 32-MeV α-particles
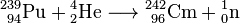 .
.
The americium-241 and curium-242 isotopes also were produced by irradiating plutonium in a nuclear reactor. The latter element was named after Marie Curie and her husband Pierre who are noted for discovering radium and for their work in radioactivity.
Bombarding curium-242 with α-particles resulted in an isotope of californium 245Cf (1950), and a similar procedure yielded in 1949 berkelium-243 from americium-241. The new elements were named after Berkeley, California, by analogy with its lanthanide homologue terbium, which was named after the village of Ytterby in Sweden.
In 1945, B. B. Cunningham obtained the first bulk chemical compound of a transplutonium element, namely americium hydroxide. Over the next three to four years, milligram quantities of americium and microgram amounts of curium were accumulated that allowed production of isotopes of berkelium (Thomson, 1949) and californium (Thomson, 1950). Sizeable amounts of these elements were produced only in 1958 (Burris B. Cunningham and Stanley G. Thomson), and the first californium compound (0.3 µg of CfOCl) was obtained only in 1960 by B. B. Cunningham and J. C. Wallmann).
Einsteinium and fermium were identified in 1952–1953 in the fallout from the " Ivy Mike" nuclear test (1 November 1952), the first successful test of a hydrogen bomb. Instantaneous exposure of uranium-238 to a large neutron flux resulting from the explosion produced heavy isotopes of uranium, including uranium-253 and uranium-255, and their β-decay yielded einsteinium-253 and fermium-255. The discovery of the new elements and the new data on neutron capture were initially kept secret on the orders of the U.S. military until 1955 due to Cold War tensions. Nevertheless, the Berkeley team were able to prepare einsteinium and fermium by civilian means, through the neutron bombardment of plutonium-239, and published this work in 1954 with the disclaimer that it was not the first studies that had been carried out on the elements. The "Ivy Mike" studies were declassified and published in 1955. The first significant (submicrograms) amounts of einsteinium were produced in 1961 by Cunningham and colleagues, but this has not been done for fermium yet.
The first isotope of mendelevium, 256Md (half-life 87 min), was synthesized by Albert Ghiorso, Glenn T. Seaborg, Gregory R. Choppin, Bernard G. Harvey and Stanley G. Thompson when they bombarded an 253Es target with alpha particles in the 60-inch cyclotron of Berkeley Radiation Laboratory; this was the first isotope of any element to be synthesized one atom at a time.
There were several attempts to obtain isotopes of nobelium by Swedish (1957) and American (1958) groups, but the first reliable results was the synthesis of 256No by the Russian group ( Georgy Flyorov et al.) in 1965, as acknowledges by the IUPAC in 1992. In their experiments, Flyorov et al. bombarded uranium-238 with neon-22.
In 1961, Ghiorso et al. obtained the first isotope of lawrencium by irradiating californium (mostly californium-252) with boron-10 and boron-11 ions. The mass number of this isotope was not clearly established (possibly 258 or 259) at the time. In 1965, 256Lr were synthesized by Flyorov et al. from 243Am and 18O. Thus IUPAC recognized the nuclear physics teams at Dubna and Berkeley as the co-discoverers of lawrencium.
Isotopes
| Isotope | Half-life | Probability of spontaneous fission in % |
Emission energy, MeV (yield in %) | Specific activity of | ||
|---|---|---|---|---|---|---|
| α | γ | α, β-particles, Bq/kg | fission, Bq/kg | |||
| 241Am | 432.2(7) years | 4.3(18)×10−10 | 5.485 (84.8) 5.442 (13.1) 5.388 (1.66) |
0.059 (35.9) 0.026 (2.27) |
1.27×1014 | 546.1 |
| 243Am | 7.37(4)×103 years | 3.7(2)×10−9 | 5.275 (87.1) 5.233 (11.2) 5.181 (1.36) |
0.074 (67.2) 0.043 (5.9) |
7.39×1012 | 273.3 |
| 242Cm | 162.8(2) days | 6.2(3)×10−6 | 6.069 (25.92) 6.112 (74.08) |
0.044 (0.04) 0.102 (4×10−3) |
1.23×1017 | 7.6×109 |
| 244Cm | 18.10(2) years | 1.37(3)×10−4 | 5.762 (23.6) 5.804 (76.4) |
0.043 (0.02) 0.100 (1.5×10−3) |
2.96×1015 | 4.1×109 |
| 245Cm | 8.5(1)×103 years | 6.1(9)×10−7 | 5.529 (0.58) 5.488 (0.83) 5.361 (93.2) |
0.175 (9.88) 0.133 (2.83) |
6.35×1012 | 3.9×104 |
| 246Cm | 4.76(4)×103 years | 0.02615(7) | 5.343 (17.8) 5.386 (82.2) |
0.045 (19) | 1.13×1013 | 2.95×109 |
| 247Cm | 1.56(5)×107 years | — | 5.267 (13.8) 5.212 (5.7) 5.147 (1.2) |
0.402 (72) 0.278 (3.4) |
3.43×109 | — |
| 248Cm | 3.48(6)×105 years | 8.39(16) | 5.034 (16.52) 5.078 (75) |
— | 1.40×1011 | 1.29×1010 |
| 249Bk | 330(4) days | 4.7(2)×10−8 | 5.406 (1×10−3) 5.378 (2.6×10−4) |
0.32 (5.8×10−5) | 5.88×1016 | 2.76×107 |
| 249Cf | 351(2) years | 5.0(4)×10−7 | 6.193 (2.46) 6.139 (1.33) 5.946 (3.33) |
0.388 (66) 0.333 (14.6) |
1.51×1014 | 7.57×105 |
| 250Cf | 13.08(9) years | 0.077(3) | 5.988 (14.99) 6.030 (84.6) |
0.043 | 4.04×1015 | 3.11×1012 |
| 251Cf | 900(40) years | ? | 6.078 (2.6) 5.567 (0.9) 5.569 (0.9) |
0.177 (17.3) 0.227 (6.8) |
5.86×1013 | — |
| 252Cf | 2.645(8) years | 3.092(8) | 6.075 (15.2) 6.118 (81.6) |
0.042 (1.4×10−2) 0.100 (1.3×10−2) |
1.92×1016 | 6.14×1014 |
| 254Cf | 60.5(2) days | ≈100 | 5.834 (0.26) 5.792 (5.3×10−2) |
— | 9.75×1014 | 3.13×1017 |
| 253Es | 20.47(3) days | 8.7(3)×10−6 | 6.540 (0.85) 6.552 (0.71) 6.590 (6.6) |
0.387 (0.05) 0.429 (8×10−3) |
9.33×1017 | 8.12×1010 |
| 254Es | 275.7(5) days | < 3×10−6 | 6.347 (0.75) 6.358 (2.6) 6.415 (1.8) |
0.042 (100) 0.034 (30) |
6.9×1016 | — |
| 255Es | 39.8(12) days | 0.0041(2) | 6.267 (0.78) 6.401 (7) |
— | 4.38×1017(β) 3.81×1016(α) |
1.95×1013 |
| 255Fm | 20.07(7) hours | 2.4(10)×10−5 | 7.022 (93.4) 6.963 (5.04) 6.892 (0.62) |
0.00057 (19.1) 0.081 (1) |
2.27×1019 | 5.44×1012 |
| 256Fm | 157.6(13) min | 91.9(3) | 6.872 (1.2) 6.917 (6.9) |
— | 1.58×1020 | 1.4×1019 |
| 257Fm | 100.5(2) days | 0.210(4) | 6.752 (0.58) 6.695 (3.39) 6.622 (0.6) |
0.241 (11) 0.179 (8.7) |
1.87×1017 | 3.93×1014 |
| 256Md | 77(2) min | — | 7.142 (1.84) 7.206 (5.9) |
— | 3.53×1020 | — |
| 257Md | 5.52(5) hours | — | 7.074 (14) | 0.371 (11.7) 0.325 (2.5) |
8.17×1019 | — |
| 258Md | 51.5(3) days | — | 6.73 | — | 3.64×1017 | — |
| 255No | 3.1(2) min | — | 8.312 (1.16) 8.266 (2.6) 8.121 (27.8) |
0.187 (3.4) | 8.78×1021 | — |
| 259No | 58(5) min | — | 7.455 (9.8) 7.500 (29.3) 7.533 (17.3) |
— | 4.63×1020 | — |
| 256Lr | 27(3) s | < 0.03 | 8.319 (5.4) 8.390 (16) 8.430 (33) |
— | 5.96×1022 | — |
| 257Lr | 646(25) ms | — | 8.796 (18) 8.861 (82) |
— | 1.54×1024 | — |

Thirty-one isotopes of actinium and eight excited isomeric states of some of its nuclides were identified by 2010. Three isotopes, 225Ac, 227Ac and 228Ac, were found in nature and the others were produced in the laboratory; only the three natural isotopes are used in applications. Actinium-225 is a member of radioactive neptunium series; it was first discovered in 1947 as a fission product of uranium-233, it is an α-emitter with a half-life of 10 days. Actinium-225 is less available than actinium-228, but is more promising in radiotracer applications. Actinium-227 (half-life 21.77 years) occurs in all uranium ores, but in small quantities. One gram of uranium (in radioactive equilibrium) contains only 2×10−10 gram of 227Ac. Actinium-228 is a member of radioactive thorium series formed by the decay of 228Ra; it is a β– emitter with a half-life of 6.15 hours. In one tonne of thorium there is 5×10−8 gram of 228Ac. It was discovered by Otto Hahn in 1906.
Twenty nine isotopes of protactinium are known with mass numbers 212–240 as well as three excited isomeric states. Only 231Pa and 234Pa have been found in nature. All the isotopes have short lifetime, except for protactinium-231 (half-life 32,760 years). The most important isotopes are 231Pa and 233Pa, which is an intermediate product in obtaining uranium-233 and is the most affordable among artificial isotopes of protactinium. 233Pa has convenient half-life and energy of γ-radiation, and thus was used in most studies of protactinium chemistry. Protactinium-233 is a β-emitter with a half-life of 26.97 days.
Uranium has the highest number (25) of both natural and synthetic isotopes. They have mass numbers of 217–242, and three of them, 234U, 235U and 238U, are present in appreciable quantities in nature. Among others, the most important is 233U, which is a final product of transformations of 232Th irradiated by slow neutrons. 233U has a very higher fission efficiency by low-energy (thermal) neutrons, compared e.g. with 235U. Most uranium chemistry studies were carried out on uranium-238 owing to its long half-life of 4.4×109 years.
There are 19 isotopes of neptunium with mass numbers from 225 to 244; they are all highly radioactive. The most popular among scientists are long-lived 237Np (t½ = 2.20×106 years) and short-lived 239Np, 238Np (t½ ~ 2 days).
Sixteen isotopes of americium are known with mass numbers from 232 to 248. The most important are 241Am and 243Am, which are alpha-emitters and also emit soft, but intense γ-rays; both of them can be obtained in an isotopically pure form. Chemical properties of americium were first studied with 241Am, but later shifted to 243Am, which is almost 20 times less radioactive. The disadvantage of 243Am is production of the short-lived daughter isotope 239Np, which has to be considered in the data analysis.
Among 19 isotopes of curium, the most accessible are 242Cm and 244Cm; they are α-emitters, but with much shorter lifetime than the americium isotopes. These isotopes emit almost no γ-radiation, but undergo spontaneous fission with the associated emission of neutrons. More long-lived isotopes of curium (245–248Cm, all α-emitters) are formed as a mixture during neutron irradiation of plutonium or americium. Upon short irradiation, this mixture is dominated by curium-246, and then curium-248 begins to accumulate. Both of these isotopes, especially 248Cm, have a longer half-life (3.48×105 years) and are much more convenient for carrying out chemical research than 242Cm and 244Cm, but they also have a rather high rate of spontaneous fission. 247Cm has the longest lifetime among isotopes of curium (1.56×107 years), but is not formed in large quantities because of the strong fission induced by thermal neutrons.
Fourteen isotopes of berkelium were identified with mass numbers 238–252. Only 249Bk is available in large quantities; it has a relatively short half-life of 330 days and emits mostly soft β-particles, which are inconvenient for detection. Its alpha radiation is rather weak (1.45×10−3% with respect to β-radiation), but is sometimes used to detect this isotope. 247Bk is an alpha-emitter with a long half-life of 1,380 years, but it is hard to obtain in appreciable quantities; it is not formed upon neutron irradiation of plutonium because of the β-stability of isotopes of curium isotopes with mass number below 248.
Isotopes of californium with mass numbers 237–256 are formed in nuclear reactors; californium-253 is a β-emitter and the rest are α-emitters. The isotopes with even mass numbers (250Cf, 252Cf and 254Cf) have a high rate of spontaneous fission, especially 254Cf of which 99.7% decays by spontaneous fission. Californium-249 has a relatively long half-life (352 years), weak spontaneous fission and strong γ-emission that facilitates its identification. 249Cf is not formed in large quantities in a nuclear reactor because of the slow β-decay of the parent isotope 249Bk and a large cross section of interaction with neutrons, but it can be accumulated in the isotopically pure form as the β-decay product of (pre-selected) 249Bk. Californium produced by reactor-irradiation of plutonium mostly consists of 250Cf and 252Cf, the latter being predominant for large neutron fluences, and its study is hindered by the strong neutron radiation.
| Parent isotope |
t½ | Daughter isotope |
t½ | Time to establish radioactive equilibrium |
|---|---|---|---|---|
| 243Am | 7370 years | 239Np | 2.35 days | 47.3 days |
| 245Cm | 8265 years | 241Pu | 14 years | 129 years |
| 247Cm | 1.64×107 years | 243Pu | 4.95 hours | 7.2 days |
| 254Es | 270 days | 250Bk | 3.2 hours | 35.2 hours |
| 255Es | 39.8 days | 255Fm | 22 hours | 5 days |
| 257Fm | 79 days | 253Cf | 17.6 days | 49 days |
Among the 16 known isotopes of einsteinium with mass numbers from 241 to 257 the most affordable is 253Es. It is an α-emitter with a half-life of 20.47 days, a relatively weak γ-emission and small spontaneous fission rate as compared with the isotopes of californium. Prolonged neutron irradiation also produces a long-lived isotope 254Es (t½ = 275.5 days).
Nineteen isotopes of fermium are known with mass numbers of 242–260. 254Fm, 255Fm and 256Fm are α-emitters with a short half-life (hours), which can be isolated in significant amounts. 257Fm (t½ = 100 days) can accumulate upon prolonged and strong irradiation. All these isotopes are characterized by high rates of spontaneous fission.
Among the 15 known isotopes of mendelevium (mass numbers from 245 to 260), the most studied is 256Md, which mainly decays through the electron capture (α-radiation is ≈10%) with the half-life of 77 minutes. Another alpha emitter, 258Md, has a half-life of 53 days. Both these isotopes are produced from rare einsteinium (253Es and 255Es respectively), that limits their so their availability.
Long-lived isotopes of nobelium and isotopes of lawrencium (and of heavier elements) have relatively small half-lives. For nobelium 11 isotopes are known with mass numbers 250–260 and 262. Chemical properties of nobelium and lawrencium were studied with 255No (t½ = 3 min) and 256Lr (t½ = 35 s). The longest-lived nobelium isotope 259No has a half-life of 1.5 hours.
Distribution in nature

Thorium and uranium are the most abundant actinides in nature with the respective mass concentrations of 1.6×10−3% and 4×10−4%. Uranium mostly occurs in the Earth's crust as a mixture of its oxides in the minerals uraninite, which is also called pitchblende because of its black colour. There are several dozens of other uranium minerals such as carnotite (KUO2VO4·3H2O) and autunite (Ca(UO2)2(PO4)2·nH2O). The isotopic composition of natural uranium is 238U (relative abundance 99.2742%), 235U (0.7204%) and 234U (0.0054%); of these 238U has the largest half-life of 4.51×109 years. The worldwide production of uranium in 2009 amounted to 50,572 tonnes, of which 27.3% was mined in Kazakhstan. Other important uranium mining countries are Canada (20.1%), Australia (15.7%), Namibia (9.1%), Russia (7.0%), and Niger (6.4%).
| Ore | Location | Uranium content, % |
Mass ratio 239Pu/ore |
Ratio 239Pu/U (×1012) |
|---|---|---|---|---|
| Uraninite | Canada | 13.5 | 9.1×10−12 | 7.1 |
| Uraninite | Congo | 38 | 4.8×10−12 | 12 |
| Uraninite | Colorado, US | 50 | 3.8×10−12 | 7.7 |
| Monazite | Brazil | 0.24 | 2.1×10−14 | 8.3 |
| Monazite | North Carolina, US | 1.64 | 5.9×10−14 | 3.6 |
| Fergusonite | - | 0.25 | <1×10−14 | <4 |
| Carnotite | - | 10 | <4×10−14 | <0.4 |
The most abundant thorium minerals are thorianite (ThO2), thorite (ThSiO4) and monazite, ((Th,Ca,Ce)PO4). Most thorium minerals contain uranium and vice versa; and they all have significant fraction of lanthanides. Rich deposits of thorium minerals are located in the United States (440,000 tonnes), Australia and India (~300,000 tonnes each) and Canada (~100,000 tonnes).
The abundance of actinium in the Earth's crust is only about 5×10−15%. Actinium is mostly present in uranium-containing, but also in other minerals, though in much smaller quantities. The content of actinium in most natural objects corresponds to the isotopic equilibrium of parent isotope 235U, and it is not affected by the weak Ac migration. Protactinium is more abundant (10−12%) in the Earth's crust than actinium. It was discovered in the uranium ore in 1913 by Fajans and Göhring. As actinium, the distribution of protactinium follows that of 235U.
The half-life of the longest-lived isotope of neptunium, 237Np, is negligible compared to the age of the Earth. Thus neptunium is present in nature in negligible amounts produced as intermediate decay products of other isotopes. Traces of plutonium in uranium minerals were first found in 1942, and the more systematic results on 239Pu are summarized in the table (no other plutonium isotopes could be detected in those samples). The upper limit of abundance of the longest-living isotope of plutonium, 244Pu, is 3×10−20%. Plutonium could not be detected in samples of lunar soil. Owing to its scarcity in nature, most plutonium is produced synthetically.
Negligible amounts of americium, curium, berkelium and californium are produced by neutron capture reactions and beta decay in very highly concentrated uranium-bearing deposits.
Extraction

Owing to the low abundance of actinides, their extraction is a complex, multistep process. Fluorides of actinides are usually used because they are insoluble in water and can be easily separated with redox reactions. Fluorides are reduced with calcium, magnesium or barium:
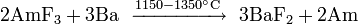
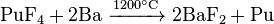
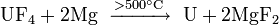
Among the actinides, thorium and uranium are the easiest to isolate. Thorium is extracted mostly from monazite: thorium diphosphate (Th(PO4)2) is reacted with nitric acid, and the produced thorium nitrate treated with tributyl phosphate. Rare-earth impurities are separated by increasing the pH in sulfate solution.
In another extraction method, monazite is decomposed with a 45% aqueous solution of sodium hydroxide at 140 °C. Mixed metal hydroxides are extracted first, filtered at 80 °C, washed with water and dissolved with concentrated hydrochloric acid. Next, the acidic solution is neutralized with hydroxides to pH = 5.8 that results in precipitation of thorium hydroxide (Th(OH)4) contaminated with ~3% of rare-earth hydroxides; the rest of rare-earth hydroxides remains in solution. Thorium hydroxide is dissolved in an inorganic acid and then purified from the rare earth elements. An efficient method is the dissolution of thorium hydroxide in nitric acid, because the resulting solution can be purified by extraction with organic solvents:

Th(OH)4 + 4 HNO3 → Th(NO3)4 + 4 H2O
Metallic thorium is separated from the anhydrous oxide, chloride or fluoride by reacting it with calcium in an inert atmosphere:
ThO2 + 2 Ca → 2 CaO + Th
Sometimes thorium is extracted by electrolysis of a fluoride in a mixture of sodium and potassium chloride at 700–800 °C in a graphite crucible. Highly pure thorium can be extracted from its iodide with the crystal bar process.
Uranium is extracted from its ores in various ways. In one method, the ore is burned and then reacted with nitric acid to convert uranium into a dissolved state. Treating the solution with a solution of tributyl phosphate (TBP) in kerosene transforms uranium into an organic form UO2(NO3)2(TBP)2. The insoluble impurities are filtered and the uranium is extracted by reaction with hydroxides as (NH4)2U2O7 or with hydrogen peroxide as UO4·2H2O.
When the uranium ore is rich in such minerals as dolomite, magnesite, etc., those minerals consume much acid. In this case, the carbonate method is used for uranium extraction. Its main component is an aqueous solution of sodium carbonate, which converts uranium into a complex [UO2(CO3)3]4–, which is stable in aqueous solutions at low concentrations of hydroxide ions. The advantages of the sodium carbonate method are that the chemicals have low corrosivity (compared to nitrates) and that most non-uranium metals precipitate from the solution. The disadvantage is that tetravalent uranium compounds precipitate as well. Therefore, the uranium ore is treated with sodium carbonate at elevated temperature and under oxygen pressure:
- 2 UO2 + O2 + 6 CO2−
3 → 2 [UO2(CO3)3]4–
This equation suggests that the best solvent for the uranium carbonate processing is a mixture of carbonate with bicarbonate. At high pH, this results in precipitation of diuranate, which is treated with hydrogen in the presence of nickel yielding an insoluble uranium tetracarbonate.
Another separation method uses polymeric resins as a polyelectrolyte. Ion exchange processes in the resins result in separation of uranium. Uranium from resins is washed with a solution of ammonium nitrate or nitric acid that yields uranyl nitrate, UO2(NO3)2·6H2O. When heated, it turns into UO3, which is converted to UO2 with hydrogen:
- UO3 + H2 → UO2 + H2O
Reacting uranium dioxide with fluoric acid changes it to uranium tetrafluoride, which yields uranium metal upon reaction with magnesium metal:
- 4 HF + UO2 → UF4 + 2 H2O
To extract plutonium, neutron-irradiated uranium is dissolved in nitric acid, and a reducing agent ( FeSO4, or H2O2) is added to the resulting solution. This addition changes the oxidation state of plutonium from +6 to +4, while uranium remains in the form of uranyl nitrate (UO2(NO3)2). The solution is treated with a reducing agent and neutralized with ammonium carbonate to pH = 8 that results in precipitation of Pu4+ compounds.
In another method, Pu4+and UO2+
2 are first extracted with tributyl phosphate, then reacted with hydrazine washing out the recovered plutonium.
The major difficulty in separation of actinium is the similarity of its properties with those of lanthanum. Thus actinium is either synthesized in nuclear reactions from isotopes of radium or separated using ion-exchange procedures.
Properties
Actinides have similar properties to lanthanides. The 6 d and 7 s electronic shells are filled in actinium and thorium, and the 5 f shell is being filled with further increase in atomic number; the 4f shell is filled in the lanthanides. The first experimental evidence for the filling of the 5f shell in actinides was obtained by McMillan and Abelson in 1940. As in lanthanides (see lanthanide contraction), the ionic radius of actinides monotonically decreases with atomic number (see also Aufbau principle).
| Property | Ac | Th | Pa | U | Np | Pu | Am | Cm | Bk | Cf | Es | Fm | Md | No | Lr |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Core charge | 89 | 90 | 91 | 92 | 93 | 94 | 95 | 96 | 97 | 98 | 99 | 100 | 101 | 102 | 103 |
| atomic mass | [227] | 232.0381 | 231.03588 | 238.02891 | [237] | [244] | [243] | [247] | [247] | [251] | [252] | [257] | [258] | [259] | [262] |
| Number of natural isotopes | 3 | 9 | 5 | 9 | 4 | 5 | 5 | 8 | 2 | 5 | — | — | — | — | — |
| Natural isotopes | 225, 227–228 | 226–232, 234–235 | 231, 233–236 | 232–240 | 237–240 | 238–240, 242, 244 | 241–245 | 242–249 | 249–250 | 249–253 | — | — | — | — | — |
| Longest-lived isotope | 227 | 232 | 231 | 238 | 237 | 244 | 243 | 247 | 247 | 251 | 252 | 257 | 258 | 259 | 262 |
| Half-life of the longest-lived isotope | 21.8 years | 14 billion years | 32,500 years | 4.47 billion years | 2.14 million years | 80.8 million years | 7,370 years | 15.6 million years | 1,400 years | 900 years | 1.29 years | 100.5 days | 52 days | 58 min | 261 min |
| Electronic configuration in the ground state | 6d17s2 | 6d27s2 | 5f26d17s2or 5f16d27s2 | 5f36d17s2 | 5f46d17s2or 5f57s2 | 5f67s2 | 5f77s2 | 5f76d17s2 | 5f97s2or 5f86d17s2 | 5f107s2 | 5f117s2 | 5f127s2 | 5f137s2 | 5f147s2 | 5f147s27p1 |
| Oxidation state | 3 | 3, 4 | 3, 4, 5 | 3, 4, 5, 6 | 3, 4, 5, 6, 7 | 3, 4, 5, 6, 7 | 2, 3, 4 | 3, 4 | 3, 4 | 2, 3 | 2, 3 | 2, 3 | 2, 3 | 2, 3 | 3 |
| Metallic radius, nm | 0.203 | 0.180 | 0.162 | 0.153 | 0.150 | 0.162 | 0.173 | 0.174 | 0.170 | 0.186 | 0.186 | — | — | — | — |
| Ionic radius, nm: An4+ An3+ |
— 0.126 |
0.114 — |
0.104 0.118 |
0.103 0.118 |
0.101 0.116 |
0.100 0.115 |
0.099 0.114 |
0.099 0.112 |
0.097 0.110 |
0.096 0.109 |
0.085 0.098 |
0.084 0.091 |
0.084 0.090 |
0.084 0.095 |
0.083 0.088 |
| Temperature, °C: melting boiling |
1050 3300 |
1750 4800 |
1572 4400 |
1130 3800 |
640 3900 |
640 3230 |
1176 2610 |
1340 — |
1050 — |
900 — |
860 — |
1530 — |
830 — |
830 — |
1630 — |
| Density, g/cm3 | 10.07 | 11.78 | 15.37 | 19.06 | 20.25 | 19.84 | 11.7 | 13.51 | 14.78 | 15.1 | 8.84 | ||||
| Standard electrode potential, V: E° (An4+/An0) E° (An3+/An0) |
— −2.13 |
−1.83 — |
−1.47 — |
−1.38 −1.66 |
−1.30 −1.79 |
−1.25 −2.00 |
−0.90 −2.07 |
−0.75 −2.06 |
−0.55 −1.96 |
−0.59 −1.97 |
−0.36 −1.98 |
−0.29 −1.96 |
— −1.74 |
— −1.20 |
- −2.10 |
| Colour [M(H2O)n]4+ [M(H2O)n]3+ |
— Colorless |
Colorless Blue |
Yellow Dark blue |
Green Purple |
Yellow-green Purple |
Brown Violet |
Red Rose |
Yellow Colorless |
Beige Yellow-green |
Green Green |
— Pink |
— — |
— — |
— — |
— — |
| Oxidation state | 89 | 90 | 91 | 92 | 93 | 94 | 95 | 96 | 97 | 98 | 99 |
| +3 | Ac3+ | Th3+ | Pa3+ | U3+ | Np3+ | Pu3+ | Am3+ | Cm3+ | Bk3+ | Cf3+ | Es3+ |
| +4 | Th4+ | Pa4+ | U4+ | Np4+ | Pu4+ | Am4+ | Cm4+ | Bk4+ | Cf4+ | ||
| +5 | PaO+ 2 |
UO+ 2 |
NpO+ 2 |
PuO+ 2 |
AmO+ 2 |
||||||
| +6 | UO2+ 2 |
NpO2+ 2 |
PuO2+ 2 |
AmO2+ 2 |
|||||||
| +7 | NpO3+ 2 |
PuO3+ 2 |
[AmO6]5- |
Physical properties
 |
 |
| Major crystal structures of some actinides vs. temperature | Metallic and ionic radii of actinides |


Actinides are typical metals. All of them are soft and have a silvery colour (but tarnish in air), relatively high density and plasticity. Some of them can be cut with a knife. Their electrical resistivity varies between 15 and 150 µOhm·cm. The hardness of thorium is similar to that of soft steel, so heated pure thorium can be rolled in sheets and pulled into wire. Thorium is nearly half as dense as uranium and plutonium, but is harder than either of them. All actinides are radioactive, paramagnetic, and, with the exception of actinium, have several crystalline phases: plutonium has seven, and uranium, neptunium and californium three. Crystal structures of protactinium, uranium, neptunium and plutonium do not have clear analogs among the lanthanides and are more similar to those of the 3d-transition metals.
All actinides are pyrophoric, especially when finely divided, that is, they spontaneously ignite upon reaction with air. The melting point of actinides does not have a clear dependence on the number of f-electrons. The unusually low melting point of neptunium and plutonium (~640 °C) is explained by hybridization of 5f and 6d orbitals and the formation of directional bonds in these metals.
| Lanthanides | Ln3+, Å | Actinides | An3+, Å | An4+, Å |
|---|---|---|---|---|
| Lanthanum | 1.061 | Actinium | 1.11 | – |
| Cerium | 1.034 | Thorium | 1.08 | 0.99 |
| Praseodymium | 1.013 | Protactinium | 1.05 | 0.93 |
| Neodymium | 0.995 | Uranium | 1.03 | 0.93 |
| Promethium | 0.979 | Neptunium | 1.01 | 0.92 |
| Samarium | 0.964 | Plutonium | 1.00 | 0.90 |
| Europium | 0.950 | Americium | 0.99 | 0.89 |
| Gadolinium | 0.938 | Curium | 0.98 | 0.88 |
| Terbium | 0.923 | Berkelium | - | - |
| Dysprosium | 0.908 | Californium | - | - |
| Holmium | 0.894 | Einsteinium | - | - |
| Erbium | 0.881 | Fermium | - | - |
| Thulium | 0.869 | Mendelevium | - | - |
| Ytterbium | 0.858 | Nobelium | - | - |
| Lutetium | 0.848 | Lawrencium | - | - |
Chemical properties
Like the lanthanides, all actinides are highly reactive with halogens and chalcogens; however, the actinides react more easily. Actinides, especially those with a small number of 5f-electrons, are prone to hybridization. This is explained by the similarity of the electron energies at the 5f, 7s and 6d shells. Most actinides exhibit a larger variety of valence states, and the most stable are +6 for uranium, +5 for protactinium and neptunium, +4 for thorium and plutonium and +3 for actinium and other actinides.
Chemically, actinium is similar to lanthanum, which is explained by their similar ionic radii and electronic structure. Like lanthanum, actinium has oxidation of +3, but it is less reactive and has more pronounced basic properties. Among other trivalent actinides Ac3+ is least acidic, i.e. has the weakest tendency to hydrolyze in aqueous solutions.
Thorium is rather active chemically. Owing to lack of electrons on 6d and 5f orbitals, the tetravalent thorium compounds are colorless. At pH < 3, the solutions of thorium salts are dominated by the cations [Th(H2O)8]4+. The Th4+ ion is relatively large, and depending on the coordination number can have a radius between 0.95 and 1.14 Å. As a result, thorium salts have a weak tendency to a hydrolysis. The distinctive ability of thorium salts is their high solubility, not only in water, but also in polar organic solvents.
Protactinium exhibits two valence states; the +5 is stable, and the +4 state easily oxidizes to protactinium(V). Thus tetravalent protactinium in solutions is obtained by the action of strong reducing agents in a hydrogen atmosphere. Tetravalent protactinium is chemically similar to uranium(IV) and thorium(IV). Fluorides, phosphates, hypophosphate, iodate and phenylarsonates of protactinium(IV) are insoluble in water and dilute acids. Protactinium forms soluble carbonates. The hydrolytic properties of pentavalent protactinium are close to those of tantalum(V) and niobium(V). The complex chemical behaviour of protactinium is a consequence of the start of the filling of the 5f shell in this element.
Uranium has a valence from 3 to 6, the last being most stable. In the hexavalent state, uranium is very similar to the sixth group elements. Many compounds of uranium(IV) and uranium(VI) are nonstoichiometric, i.e. have variable composition. For example, the actual chemical formula of uranium dioxide is UO2+x, where x varies between −0.4 and 0.32. Uranium(VI) compounds are weak oxidants. Most of them contain the linear " uranyl" group, UO2+
2. Between 4 to 6 ligands can be accommodated in an equatorial plane perpendicular to the uranyl group. The uranyl group acts as a hard acid and forms stronger complexes with oxygen-donor ligands than with nitrogen-donor ligands. NpO2+
2 and PuO2+
2 are also the common form of Np and Pu in the +6 oxidation state. Uranium(IV) compounds exhibit reducing properties, e.g., they are easily oxidized by atmospheric oxygen. Uranium(III) is a very strong reducing agent. Owing to the presence of d-shell, uranium (as well as many other actinides) forms organometallic compounds, such as UIII(C5H5)3 and UIV(C5H5)4.
Neptunium has valence states from 3 to 7, which can be simultaneously observed in solutions. The most stable state in solution is +5, but the valence +4 is preferred in solid neptunium compounds. Neptunium metal is very reactive. Ions of neptunium are prone to hydrolysis and formation of coordination compounds.
Plutonium also exhibits the valence between 3 and 7, and thus is chemically similar to neptunium and uranium. It is highly reactive, and quickly forms an oxide film in air. Plutonium reacts with hydrogen even at temperatures as low as 25–50 °C; it also easily forms halides and intermetallic compounds. Hydrolysis reactions of plutonium ions of different oxidation states are quite diverse. Plutonium(V) can enter polymerization reactions.
The largest chemical diversity among actinides is observed in americium, which can have valence between 2 and 6. Divalent americium is obtained only in dry compounds and non-aqueous solutions ( acetonitrile). Oxidation states +3, +5 and +6 are typical for aqueous solutions, but also in the solid state. Tetravalent americium forms stable solid compounds (dioxide, fluoride and hydroxide) as well as complexes in aqueous solutions. It was reported that in alkaline solution americium can be oxidized to the heptavalent state, but these data proved erroneous. The most stable valence of americium is 3 in the aqueous solutions and 3 or 4 in solid compounds.
Valence 3 is dominant in all subsequent elements up to lawrencium (with the possible exception of nobelium). Curium can be tetravalent in solids (fluoride, dioxide). Berkelium, along with a valence of +3, also shows the valence of +4, more stable than that of curium; the valence 4 is observed in solid fluoride and dioxide. The stability of Bk4+ in aqueous solution is close to that of Ce4+. Only valence 3 was observed for californium, einsteinium and fermium. The divalent state is proven for mendelevium and nobelium, and in nobelium it is more stable than the trivalent state. Lawrencium shows valence 3 both in solutions and solids.
The redox potential 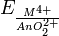 increases from −0.32 V in uranium, through 0.34 V (Np) and 1.04 V (Pu) to 1.34 V in americium revealing the increasing reduction ability of the An4+ ion from americium to uranium. All actinides form AnH3 hydrides of black colour with salt-like properties. Actinides also produce carbides with the general formula of AnC or AnC2 (U2C3 for uranium) as well as sulfides An2S3 and AnS2.
increases from −0.32 V in uranium, through 0.34 V (Np) and 1.04 V (Pu) to 1.34 V in americium revealing the increasing reduction ability of the An4+ ion from americium to uranium. All actinides form AnH3 hydrides of black colour with salt-like properties. Actinides also produce carbides with the general formula of AnC or AnC2 (U2C3 for uranium) as well as sulfides An2S3 and AnS2.
-

Uranyl nitrate (UO2(NO3)2)
-

Aqueous solutions of uranium III, IV, V, VI salts
-

Aqueous solutions of neptunium III, IV, V, VI, VII salts
-

Aqueous solutions of plutonium III, IV, V, VI, VII salts
-

Uranium tetrachloride
-

Uranium hexafluoride
-

U3O8 (yellowcake)







Compounds
Oxides and hydroxides
| Compound | Colour | Crystal symmetry, type | Lattice constants, Å | Density, g/cm3 | Temperature, °C | ||
|---|---|---|---|---|---|---|---|
| a | b | c | |||||
| Ac2O3 | White | Hexagonal, La2O3 | 4.07 | - | 6.29 | 9.19 | – |
| PaO2 | - | Cubic, CaF2 | 5.505 | - | - | - | - |
| Pa2O5 | White | cubic, CaF2 Cubic Tetragonal Hexagonal Rhombohedral Orthorhombic |
5.446 10.891 5.429 3.817 5.425 6.92 |
- - - - - 4.02 |
- 10.992 5.503 13.22 - 4. 18 |
- | 700 700–1100 1000 1000–1200 1240–1400 – |
| ThO2 | Colorless | Cubic | 5.59 | - | - | 9.87 | – |
| UO2 | Black-brown | Cubic | 5.47 | - | - | 10.9 | – |
| NpO2 | Greenish-brown | Cubic, CaF2 | 5.424 | - | - | 11.1 | – |
| PuO | Black | Cubic, NaCl | 4.96 | - | - | 13.9 | – |
| PuO2 | Olive green | Cubic | 5.39 | - | - | 11.44 | – |
| Am2O3 | Red-brown Red-brown |
Cubic, Mn2O3 Hexagonal, La2O3 |
11.03 3.817 |
- | - 5.971 |
10.57 11.7 |
– |
| AmO2 | Black | Cubic, CaF2 | 5.376 | - | - | - | - |
| Cm2O3 | White - - |
Cubic, Mn2O2 Hexagonal, LaCl3 Monoclinic, Sm2O3 |
11.01 3.80 14.28 |
- - 3.65 |
- 6 8.9 |
11.7 | – |
| CmO2 | Black | Cubic, CaF2 | 5.37 | - | - | - | - |
| Bk2O3 | Light brown | Cubic, Mn2O3 | 10.886 | - | - | - | - |
| BkO2 | Red-brown | Cubic, CaF2 | 5.33 | - | - | - | - |
| Cf2O3 | Colorless Yellowish - |
Cubic, Mn2O3 Monoclinic, Sm2O3 Hexagonal, La2O3 |
10.79 14.12 3.72 |
- 3.59 - |
- 8.80 5.96 |
- | - |
| CfO2 | Black | Cubic | 5.31 | - | - | - | - |
| Es2O3 | - | Cubic, Mn2O3 Monoclinic Hexagonal, La2O3 |
10.07 14.1 3.7 |
- 3.59 - |
- 8.80 6 |
- | - |
| Oxidation state | 89 | 90 | 91 | 92 | 93 | 94 | 95 | 96 | 97 | 98 | 99 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| +3 | Pu2O3 | Am2O3 | Cm2O3 | Bk2O3 | Cf2O3 | Es2O3 | |||||
| +4 | ThO2 | PaO2 | UO2 | NpO2 | PuO2 | AmO2 | CmO2 | BkO2 | CfO2 | ||
| +5 | Pa2O5 | U2O5 | Np2O5 | ||||||||
| +6 | U3O8 | ||||||||||
| UO3 |
| Chemical formula | ThO2 | PaO2 | UO2 | NpO2 | PuO2 | AmO2 | CmO2 | BkO2 | CfO2 |
| CAS-number | 1314-20-1 | 12036-03-2 | 1344-57-6 | 12035-79-9 | 12059-95-9 | 12005-67-3 | 12016-67-0 | 12010-84-3 | 12015-10-0 |
| Molar mass | 264.04 | 263.035 | 270.03 | 269.047 | 276.063 | 275.06 | 270–284** | 279.069 | 283.078 |
| Melting point | 3390 °C | 2865 °C | 2547 °C | 2400 °C | 2175 °C | ||||
| Crystal structure |  An4+: __ / O2−: __ |
||||||||
| Space group | Fm3m | ||||||||
| Coordination number | An, O[4] | ||||||||
- An – actinide
**Depending on the isotopes
Some actinides can exists in several oxide forms such as An2O3, AnO2, An2O5 and AnO3. For all actinides, oxides AnO3 are amphoteric and An2O3, AnO2 and An2O5 are basic, they easily react with water, forming bases:
- An2O3 + 3 H2O → 2 An(OH)3.
These bases are poorly soluble in water and by their activity are close to the hydroxides of rare-earth metals. The strongest base is of actinium. All compounds of actinium are colorless, except for black actinium sulfide (Ac2S3). Dioxides of tetravalent actinides crystallize in the cubic system, same as in calcium fluoride.
Thorium reacting with oxygen exclusively forms dioxide:
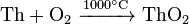
Thorium dioxide is a refractory material with the highest melting point among any known oxide (3390 °C). Adding 0.8–1% ThO2 to tungsten stabilizes its structure, so the doped filaments have better mechanical stability to vibrations. To dissolve ThO2 in acids, it is heated to 500–600 °C; heating above 600 °C produces a very resistant to acids and other reagents form of ThO2. Small addition of fluoride ions catalyses dissolution of thorium dioxide in acids.
Two protactinium oxides were obtained: PaO2 (black) and Pa2O5(white); the former is isomorphic with ThO2 and the latter is easier to obtain. Both oxides are basic, and Pa(OH)5 is a weak, poorly soluble base.
Decomposition of certain salts of uranium, for example UO2(NO3)·6H2O in air at 400 °C, yields orange or yellow UO3. This oxide is amphoteric and forms several hydroxides, the most stable being UO2(OH)2. Reaction of uranium(VI) oxide with hydrogen results in uranium dioxide, which is similar in its properties with ThO2. This oxide is also basic and corresponds to the uranium hydroxide (U(OH)4).
Plutonium, neptunium and americium form two basic oxides: An2O3 and AnO2. Neptunium trioxide is unstable; thus, only Np3O8 could be obtained so far. However, the oxides of plutonium and neptunium with the chemical formula AnO2 and An2O3 are well characterized.
Salts
| Chemical formula | AcCl3 | UCl3 | NpCl3 | PuCl3 | AmCl3 | CmCl3 | BkCl3 | CfCl3 |
| CAS-number | 22986-54-5 | 10025-93-1 | 20737-06-8 | 13569-62-5 | 13464-46-5 | 13537-20-7 | 13536-46-4 | 13536-90-8 |
| Molar mass | 333.386 | 344.387 | 343.406 | 350.32 | 349.42 | 344–358** | 353.428 | 357.438 |
| Melting point | 837 °C | 800 °C | 767 °C | 715 °C | 695 °C | 603 °C | 545 °C | |
| Boiling point | 1657 °C | 1767 °C | 850 °C | |||||
| Crystal structure |  An3+: __ / Cl−: __ |
|||||||
| Space group | P63/m | |||||||
| Coordination number | An*[9], Cl | |||||||
| Lattice constants | a = 762 pm c = 455 pm |
a = 745.2 pm c = 432.8 pm |
a = 739.4 pm c = 424.3 pm |
a = 738.2 pm c = 421.4 pm |
a = 726 pm c = 414 pm |
a = 738.2 pm c = 412.7 pm |
a = 738 pm c = 409 pm |
|
- *An – actinide
**Depending on the isotopes
| Compound | Colour | Crystal symmetry, type | Lattice constants, Å | Density, g/cm3 | ||
|---|---|---|---|---|---|---|
| a | b | c | ||||
| AcF3 | White | Hexagonal, LaF3 | 4.27 | - | 7.53 | 7.88 |
| PaF4 | Dark brown | Monoclinic | 12.7 | 10.7 | 8.42 | – |
| PaF5 | Black | Tetragonal, β-UF5 | 11.53 | - | 5.19 | – |
| ThF4 | Colorless | Monoclinic | 13 | 10.99 | 8.58 | 5.71 |
| UF3 | Reddish-purple | Hexagonal | 7.18 | - | 7.34 | 8.54 |
| UF4 | Green | Monoclinic | 11.27 | 10.75 | 8.40 | 6.72 |
| α-UF5 | Bluish | Tetragonal | 6.52 | - | 4.47 | 5.81 |
| β-UF5 | Bluish | Tetragonal | 11.47 | - | 5.20 | 6.45 |
| UF6 | Yellowish | Orthorhombic | 9.92 | 8.95 | 5.19 | 5.06 |
| NpF3 | Black or purple | Hexagonal | 7.129 | - | 7.288 | 9.12 |
| NpF4 | Light green | Monoclinic | 12.67 | 10.62 | 8.41 | 6.8 |
| NpF6 | Orange | Orthorhombic | 9.91 | 8.97 | 5.21 | 5 |
| PuF3 | Violet-blue | Trigonal | 7.09 | - | 7.25 | 9.32 |
| PuF4 | Pale brown | Monoclinic | 12.59 | 10.57 | 8.28 | 6.96 |
| PuF6 | Red-brown | Orthorhombic | 9.95 | 9.02 | 3.26 | 4.86 |
| AmF3 | Pink or light beige | hexagonal, LaF3 | 7.04 | - | 7.255 | 9.53 |
| AmF4 | Orange-red | Monoclinic | 12.53 | 10.51 | 8.20 | – |
| CmF3 | From brown to white | Hexagonal | 4.041 | - | 7.179 | 9.7 |
| CmF4 | Yellow | Monoclinic, UF4 | 12.51 | 10.51 | 8.20 | – |
| BkF3 | Yellow-green | Trigonal, LaF3 Orthorhombic, YF3 |
6.97 6.7 |
- 7.09 |
7.14 4.41 |
10.15 9.7 |
| BkF4 | - | Monoclinic, UF4 | 12.47 | 10.58 | 8.17 | – |
| CfF3 | - - |
Trigonal, LaF3 Orthorhombic, YF3 |
6. 94 6.65 |
- 7.04 |
7.10 4.39 |
– |
| CfF4 | - - |
Monoclinic, UF4 Monoclinic, UF4 |
1.242 1.233 |
1.047 1.040 |
8.126 8.113 |
– |

Actinides easily react with halogens forming salts with the formulas MX3 and MX4 (X = halogen). So the first berkelium compound, BkCl3, was synthesized in 1962 with an amount of 3 nanograms. Like the halogens of rare earth elements, actinide chlorides, bromides, and iodides are water soluble, and fluorides are insoluble. Uranium easily yields a colorless hexafluoride, which sublimates at a temperature of 56.5 °C; because of its volatility, it is used in the separation of uranium isotopes with gas centrifuge or gaseous diffusion. Actinide hexafluorides have properties close to anhydrides. They are very sensitive to moisture and hydrolyze forming AnO2F2. The pentachloride and black hexachloride of uranium were synthesized, but they are both unstable.
Action of acids on actinides yields salts, and if the acids are non-oxidizing then the actinide in the salt is in low-valence state:
- U + 2 H2SO4 → U (SO4)2 + 2 H2
- 2 Pu + 6 HCl → 2 PuCl3 + 3 H2
However, in these reactions the regenerating hydrogen can react with the metal, forming the corresponding hydride. Uranium reacts with acids and water much more easily than thorium.
Actinide salts can also be obtained by dissolving the corresponding hydroxides in acids. Nitrates, chlorides, sulfates and perchlorates of actinides are water soluble. When crystallizing from aqueous solutions, these salts forming a hydrates, such as Th(NO3)4·6H2O, Th(SO4)2·9H2O and Pu2(SO4)3·7H2O. Salts of high-valence actinides easily hydrolyze. So, colorless sulfate, chloride, perchlorate and nitrate of thorium transform into basic salts with formulas Th(OH)2SO4 and Th(OH)3NO3. The solubility and insolubility of trivalent and tetravalent actinides is like that of lanthanide salts. So phosphates, fluorides, oxalates, iodates and carbonates of actinides are weakly soluble in water; they precipitate as hydrates, such as ThF4·3H2O and Th(CrO4)2·3H2O.
Actinides with oxidation state +6, except for the AnO22+-type cations, form [AnO4]2–, [An2O7]2– and other complex anions. For example, uranium, neptunium and plutonium form salts of the Na2UO4 (uranate) and (NH4)2U2O7 (diuranate) types. In comparison with lanthanides, actinides more easily form coordination compounds, and this ability increases with the actinide valence. Trivalent actinides do not form fluoride coordination compounds, whereas tetravalent thorium forms K2ThF6, KThF5, and even K5ThF9 complexes. Thorium also forms the corresponding sulfates (for example Na2SO4·Th (SO4)2·5H2O), nitrates and thiocyanates. Salts with the general formula An2Th(NO3)6·nH2O are of coordination nature, with the coordination number of thorium equal to 12. Even easier is to produce complex salts of pentavalent and hexavalent actinides. The most stable coordination compounds of actinides – tetravalent thorium and uranium – are obtained in reactions with diketones, e.g. acetylacetone.
Applications

While actinides have some established daily-life applications, such as in smoke detectors (americium) and gas mantles (thorium), they are mostly used in nuclear weapons and use as a fuel in nuclear reactors. The last two areas exploit the property of actinides to release enormous energy in nuclear reactions, which under certain conditions may become self-sustaining chain reaction.

The most important isotope for nuclear power applications is uranium-235. It is used in the thermal reactor, and its concentration in natural uranium does not exceed 0.72%. This isotope strongly absorbs thermal neutrons releasing much energy. One fission act of 1 gram of 235U converts into about 1 MW·day. Of importance, is that 235U emits more neutrons than it absorbs; upon reaching the critical mass, 235U enters into a self-sustaining chain reaction. Typically, uranium nucleus is divided into two fragments with the release of 2–3 neutrons, for example:
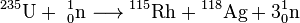
Other promising actinide isotopes for nuclear power are thorium-232 and its product from the thorium fuel cycle, uranium-233.
| Nuclear reactor |
The core of any nuclear reactor contains a set of hollow metal rods, usually made of zirconium alloys, filled with nuclear fuel cells – mostly oxide, carbide, nitride or monosulfide of uranium, plutonium or thorium, or their mixture (the so-called MOX fuel). The most common fuel is oxide of uranium-235. Fast neutrons are slowed by moderators, which contain water, carbon, deuterium, or beryllium, as thermal neutrons to increase the efficiency of their interaction with uranium-235. The rate of nuclear reaction is controlled by introducing additional rods made of boron or cadmium or a liquid absorbent, usually boric acid. Reactors for plutonium production are called breeder reactor or breeders; they have a different design and use fast neutrons. |
Emission of neutrons during the fission of uranium is important not only for maintaining the nuclear chain reaction, but also for the synthesis of the heavier actinides. Uranium-239 converts via β-decay into plutonium-239, which, like uranium-235, is capable of spontaneous fission. The world's first nuclear reactors were built not for energy, but for producing plutonium-239 for nuclear weapons.
About half of the produced thorium is used as the light-emitting material of gas mantles. Thorium is also added into multicomponent alloys of magnesium and zinc. So the Mg-Th alloys are light and strong, but also have high melting point and ductility and thus are widely used in the aviation industry and in the production of missiles. Thorium also has good electron emission properties, with long lifetime and low potential barrier for the emission. The relative content of thorium and uranium isotopes is widely used to estimate the age of various objects, including stars (see radiometric dating).
The major application of plutonium has been in nuclear weapons, where the isotope plutonium-239 was a key component due to its ease of fission and availability. Plutonium-based designs allow reducing the critical mass to about a third of that for uranium-235. The " Fat Man"-type plutonium bombs produced during the Manhattan Project used explosive compression of plutonium to obtain significantly higher densities than normal, combined with a central neutron source to begin the reaction and increase efficiency. Thus only 6.2 kg of plutonium was needed for an explosive yield equivalent to 20 kilotons of TNT. (See also Nuclear weapon design.) Hypothetically, as little as 4 kg of plutonium—and maybe even less—could be used to make a single atomic bomb using very sophisticated assembly designs.
Plutonium-238 is potentially more efficient isotope for nuclear reactors, as it has smaller critical mass than uranium-235, but releases much more thermal energy (0.56 W/g). However, its application is limited by the high price (about 1000 USD/g). This isotope has been used in thermopiles and water distillation systems of some space satellites and stations. So Galileo and Apollo spacecrafts (e.g. Apollo 14) had heaters powered by kilogram quantities of plutonium-238 oxide; this heat is also transformed into electricity with thermopiles. The decay of plutonium-238 is produced relatively harmless alpha particles and is not accompanied by gamma-irradiation. Therefore and this isotope (~160 mg) is used as the energy source in heart pacemakers where it lasts about 5 times longer than conventional batteries.
Actinium-227 is used as a neutron source. Its high specific energy (14.5 W/g) and the possibility of obtaining significant quantities of thermally stable compounds are attractive for use in long-lasting thermoelectric generators for remote use. 228Ac is used as an indicator of radioactivity in chemical research, as it emits high-energy electrons (2.18 MeV) that can be easily detected. 228Ac- 228Ra mixtures are widely used as an intense gamma-source in industry and medicine.
Development of self-glowing actinide-doped materials with durable crystalline matrices is a new area of actinide utilization as the addition of alpha-emitting radionuclides to some glasses and crystals may confer luminescence.
Toxicity


Radioactive substances can harm human health via (i) local skin contamination, (ii) internal exposure due to ingestion of radioactive isotopes, and (iii) external overexposure by β-activity and γ-radiation. Together with radium and transuranium elements, actinium is one of the most dangerous radioactive poisons with high specific α-activity. The most important feature of actinium is its ability to accumulate and remain in the surface layer of skeletons. At the initial stage of poisoning, actinium accumulates in the liver. Another danger of actinium is that it undergoes radioactive decay faster than being excreted. Adsorption from the digestive tract is much smaller (~0.05%) for actinium than radium.
Protactinium in the body tends to accumulate in the kidneys and bones. The maximum safe dose of protactinium in the human body is 0.03 µCi that corresponds to 0.5 micrograms of 231Pa. This isotope, which might be present in the air as aerosol, is 2.5×108 times more toxic than hydrocyanic acid.
Plutonium, when entering the body through air, food or blood (e.g. a wound), mostly settles in the lungs, liver and bones with only about 10% going to other organs, and remains there for decades. The long residence time of plutonium in the body is partly explained by its poor solubility in water. Some isotopes of plutonium emit ionizing α-radiation, which damages the surrounding cells. The median lethal dose (LD50) for 30 days in dogs after intravenous injection of plutonium is 0.32 milligram per kg of body mass, and thus the lethal dose for humans is approximately 22 mg for a person weighing 70 kg; the amount for respiratory exposure should be approximately four times greater. Another estimate assumes that plutonium is 50 times less toxic than radium, and thus permissible content of plutonium in the body should be 5 µg or 0.3 µCi. Such amount is nearly invisible in under microscope. After trials on animals, this maximum permissible dose was reduced to 0.65 µg or 0.04 µCi. Studies on animals also revealed that the most dangerous plutonium exposure route is through inhalation, after which 5–25% of inhaled substances is retained in the body. Depending on the particle size and solubility of the plutonium compounds, plutonium is localized either in the lungs or in the lymphatic system, or is absorbed in the blood and then transported to the liver and bones. Contamination via food is the least likely way. In this case, only about 0.05% of soluble 0.01% insoluble compounds of plutonium absorbs into blood, and the rest is excreted. Exposure of damaged skin to plutonium would retain nearly 100% of it.
Using actinides in nuclear fuel, sealed radioactive sources or advanced materials such as self-glowing crystals has many potential benefits. However, a serious concern is the extremely high radiotoxicity of actinides and their migration in the environment. Use of chemically unstable forms of actinides in MOX and sealed radioactive sources is not appropriate by modern safety standards. There is a challenge to develop stable and durable actinide-bearing materials, which provide safe storage, use and final disposal. A key need is application of actinide solid solutions in durable crystalline host phases.