Recombinant DNA technology also referred to as molecular cloning is similar to polymerase chain reaction (PCR) in that it permits the replication of a specific DNA sequence. The fundamental difference between the two methods is that molecular cloning involves replication of the DNA in a living microorganism, while PCR replicates DNA in an in vitro solution, free of living cells.
In standard molecular cloning experiments, the cloning of any DNA fragment essentially involves seven steps:
- Choice of host organism and cloning vector
- Preparation of vector DNA
- Preparation of DNA to be cloned
- Creation of recombinant DNA
- Introduction of recombinant DNA into host organism
- Selection of organisms containing recombinant DNA
- Screening for clones with desired DNA inserts and biological properties
Although a very large number of host organisms and molecular cloning vectors are in use, the great majority of molecular cloning experiments begin with a laboratory strain of the bacterium E. coli (Escherichia coli) and a plasmid cloning vector. E. coli and plasmid vectors are in common use because they are technically sophisticated, versatile, widely available, and offer rapid growth of recombinant organisms with minimal equipment. The cloning vector is treated with a restriction endonuclease to cleave the DNA at the site where foreign DNA will be inserted. The restriction enzyme is chosen to generate a configuration at the cleavage site that is compatible with that at the ends of the foreign DNA.
Typically, this is done by cleaving the vector DNA and foreign DNA with the same restriction enzyme, for example EcoRI. Most modern vectors contain a variety of convenient cleavage sites that are unique within the vector molecule (so that the vector can only be cleaved at a single site) and is located within a gene (frequently beta-galactosidase) whose inactivation can be used to distinguish recombinant from non-recombinant organisms at a later step in the process. To improve the ratio of recombinant to non-recombinant organisms, the cleaved vector may be treated with an enzyme (alkaline phosphatase) that dephosphorylates the vector ends. Vector molecules with dephosphorylated ends are unable to replicate, and replication can only be restored if foreign DNA is integrated into the cleavage site.
For cloning of genomic DNA, the DNA to be cloned is extracted from the organism of interest. Polymerase chain reaction (PCR) methods are often used for amplification of specific DNA or RNA (RT-PCR) sequences prior to molecular cloning. The purified DNA is then treated with a restriction enzyme to generate fragments with ends capable of being linked to those of the vector. If necessary, short double-stranded segments of DNA (linkers) containing desired restriction sites may be added to create end structures that are compatible with the vector. The creation of recombinant DNA is in many ways the simplest step of the molecular cloning process. DNA prepared from the vector and foreign source are simply mixed together at appropriate concentrations and exposed to an enzyme (DNA ligase) that covalently links the ends together. This joining reaction is often termed ligation. The resulting DNA mixture containing randomly joined ends is then ready for introduction into the host organism. The DNA mixture, previously manipulated in vitro, is moved back into a living cell, referred to as the host organism. The methods used to get DNA into cells are varied, and the name applied to this step in the molecular cloning process will often depend upon the experimental method that is chosen (e.g. transformation, transduction, transfection, electroporation).
When microorganisms are able to take up and replicate DNA from their local environment, the process is termed transformation, and cells that are in a physiological state such that they can take up DNA are said to be competent. When bacterial cells are used as host organisms, the selectable marker is usually a gene that confers resistance to an antibiotic that would otherwise kill the cells, typically ampicillin. Cells harboring the vector will survive when exposed to the antibiotic, while those that have failed to take up vector sequences will die. Modern bacterial cloning vectors (e.g. pUC19) use the blue-white screening system to distinguish colonies (clones) of transgenic cells from those that contain the parental vector.
In these vectors, foreign DNA is inserted into a sequence that encodes an essential part of beta-galactosidase, an enzyme whose activity results in formation of a blue-colored colony on the culture medium that is used for this work. Insertion of the foreign DNA into the beta-galactosidase coding sequence disables the function of the enzyme, so that colonies containing recombinant plasmids remain colorless (white). Therefore, recombinant clones are easily identified .
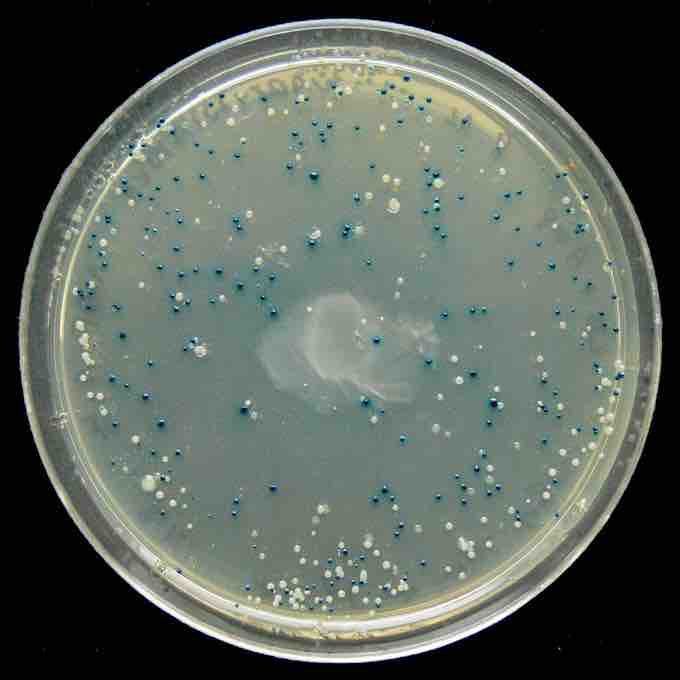
Blue White screen
The blue-white screen is a screening technique that allows for the detection of successful ligations in vector-based gene cloning. DNA of interest is ligated into a vector. The vector is then transformed into competent cell (bacteria). The competent cells are grown in the presence of X-gal. If the ligation was successful, the bacterial colony will be white; if not, the colony will be blue. This technique allows for the quick and easy detection of successful ligation.