Polycystic kidney disease (PKD or PCKD, also known as polycystic kidney syndrome) is a cystic genetic disorder of the kidneys. There are two types of PKD: autosomal dominant polycystic kidney disease (ADPKD), and the less-common autosomal recessive polycystic kidney disease (ARPKD). PKD is characterized by the presence of multiple cysts (hence, "polycystic"), typically in both kidneys. The cysts are numerous and are fluid-filled, resulting in massive enlargement of the kidneys. The disease can also damage the liver, pancreas, and, in some rare cases, the heart and brain. The two major forms of polycystic kidney disease are distinguished by their patterns of inheritance. Polycystic kidney disease is one of the most common life-threatening genetic diseases, affecting an estimated 12.5 million people worldwide.
Autosomal dominant polycystic kidney disease (ADPKD) is the most common of all the hereditary cystic kidney diseases, with an incidence of 1:1,000 to 2:1,000 live births. Studies show that 10% of end-stage renal disease (ESRD) patients treated with hemodialysis in Europe and the U.S. were initially diagnosed and treated for ADPKD. ADPKD does not appear to demonstrate a preference for any particular ethnicity.
ADPKD is characterized by progressive cyst development and bilaterally enlarged kidneys with multiple cysts. There are three genetic mutations in the PKD-1, PKD-2, and PKD3 gene with similar phenotypical presentations. Gene PKD-1 is located on chromosome 16, and codes for a protein involved in regulation of cell cycle and intracellular calcium transport in epithelial cells; it is responsible for 85% of the cases of ADPKD. PKD-2, on chromosome 4, codes for a group of voltage-linked calcium channels. PKD3 recently appeared in research papers as a postulated 3rd gene. At this time, PKD3 has not been proven. Fewer than 10% of cases of ADPKD appear in non-ADPKD families.
Cyst formation begins in utero from any point along the nephron, although fewer than 5% of nephrons are thought to be involved. As the cysts accumulate fluid, they enlarge, separate entirely from the nephron, compress the neighboring renal parenchyma, and progressively compromise renal function.
Under the function of gene defect, epithelial cells of renal tubule turn into epithelial cells of cyst wall after phenotype change and begin to have the function of secreting cyst fluid, which leads to continuous cysts enlargement.
Studies show that the incidence of autosomal recessive polycystic kidney disease (ARPKD) is 1:20,000 live births, and is typically identified in the first few weeks after birth. Unfortunately, resulting hypoplasia results in a 30% death rate in neonates with ARPKD. In ARPKD, kidneys retain their shape, but are larger than the normal anatomical range with dilated collecting ducts from the medulla to the cortex.
The major extrarenal complications of ADPKD include cerebral aneurysms, hepatic cysts, pancreatic cysts, cardiac valve disease (especially mitral valve prolapse), colonic diverticula, and aortic root dilatation.
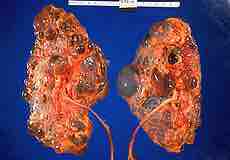
Polycystic Kidney Disease
Polycystic kidney disease, or PKD, is a cystic genetic disorder of the kidneys