Phenylketonuria (PKU) is an autosomal recessive metabolic genetic disorder characterized by a mutation in the gene for the hepatic enzyme phenylalanine hydroxylase (PAH), rendering it nonfunctional. Other non-PAH mutations can also cause PKU. This is an example of non-allelic genetic heterogeneity. The PAH gene is located on chromosome 12 in the bands 12q22-q24.1. More than 400 disease-causing mutations have been found in the PAH gene. PAH deficiency causes a spectrum of disorders, including classic phenylketonuria and hyperphenylalaninemia (a less severe accumulation of phenylalanine). Because PKU is an autosomal recessive genetic disorder, both parents must have at least one mutated allele of the PAH gene. The child must inherit both mutated alleles, one from each parent. Therefore, it is possible for a parent with the disease to have a child without it if the other parent possesses one functional allele of the gene for PAH. Yet, a child from two parents with PKU will inherit two mutated alleles every time, and therefore will also inherit the disease .
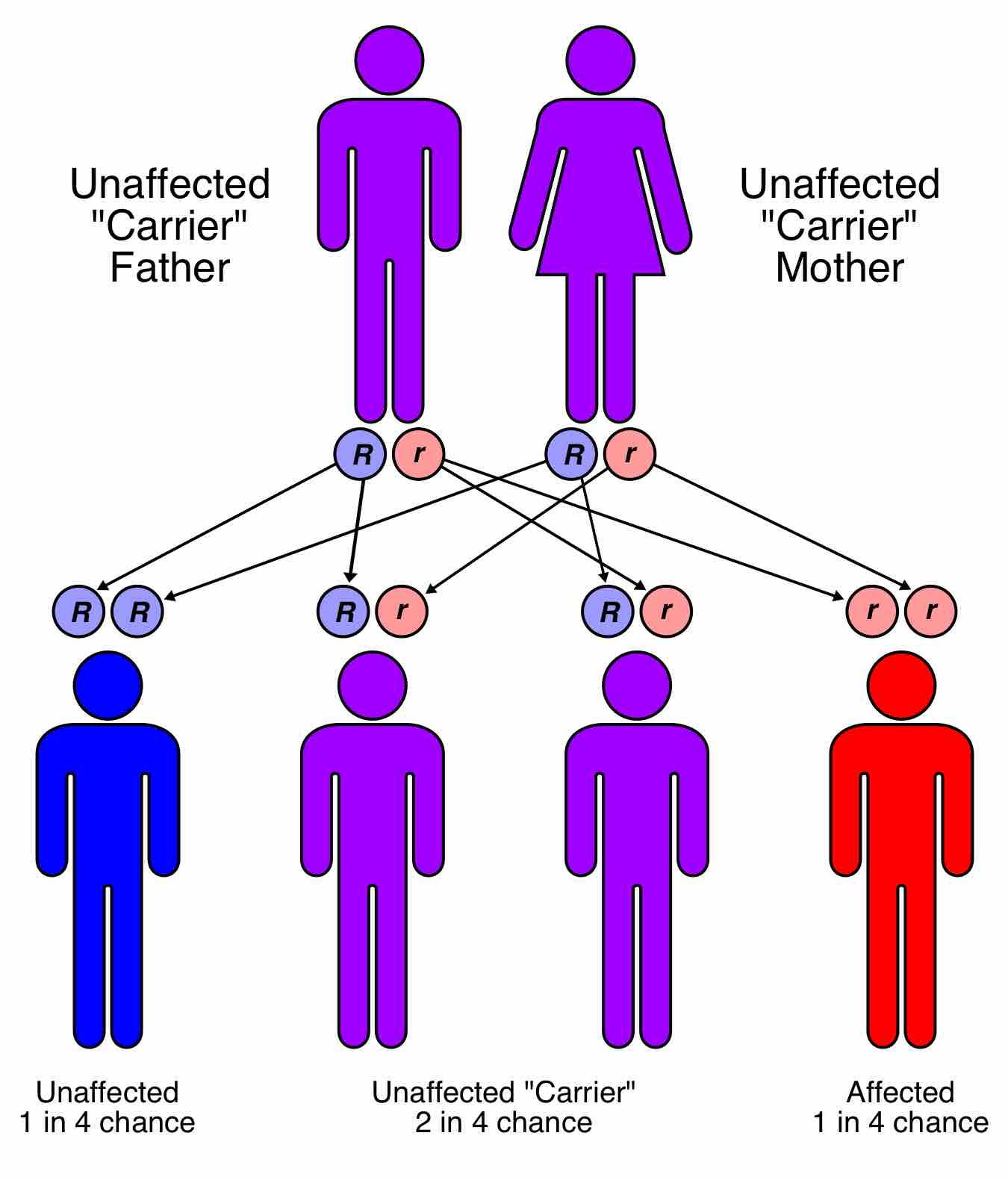
Inheritance of PKU
Phenylketonuria is inherited in an autosomal recessive fashion.
PAH enzyme is necessary for the metabolism of the amino acid phenylalanine (Phe) to the amino acid tyrosine. When PAH activity is reduced, phenylalanine accumulates. Excessive phenylalanine can be metabolized into phenylketones through the minor route, a transaminase pathway with glutamate. Metabolites include phenylacetate, phenylpyruvate, and phenethylamine. Elevated levels of phenylalanine in the blood and detection of phenylketones in the urine is diagnostic.
Phenylalanine is a large, neutral amino acid (LNAA). LNAAs compete for transport across the blood–brain barrier (BBB) via the large neutral amino acid transporter (LNAAT). If phenylalanine is in excess in the blood, it will saturate the transporter. Excessive levels of phenylalanine tend to decrease the levels of other LNAAs in the brain. However, as these amino acids are necessary for protein and neurotransmitter synthesis, Phe buildup hinders the development of the brain, causing mental retardation.
The mainstream treatment for classic PKU patients is a strict PHE-restricted diet supplemented by a medical formula containing amino acids and other nutrients. In the United States, the current recommendation is that the PKU diet should be maintained for life. Patients who are diagnosed early and maintain a strict diet can have a normal life span with normal mental development. However, recent research suggests that neurocognitive, psychosocial, quality of life, growth, nutrition, bone pathology are slightly suboptimal.
PKU is commonly included in the newborn screening panel of most countries, with varied detection techniques. Most babies in developed countries are screened for PKU soon after birth. Screening for PKU is done with bacterial inhibition assay (Guthrie test), immunoassays using fluorometric or photometric detection, or amino acid measurement using tandem mass spectrometry (MS/MS). Measurements done using MS/MS determine the concentration of Phe and the ratio of Phe to tyrosine, both of which will be elevated in PKU.
A rarer form of hyperphenylalaninemia occurs when PAH is normal, but there is a defect in the biosynthesis or recycling of the cofactor tetrahydrobiopterin (BH4) by the patient. This cofactor is necessary for proper activity of the enzyme. The coenzyme (called biopterin) can be supplemented as treatment. Those who suffer from PKU must be supplemented with tyrosine to account for PAH deficiency in converting phenylalanine to tyrosine sufficiently. Dihydrobiopterin reductase activity is to replenish quinonoid-dihydrobiopterin back into its tetrahydrobiopterin form, which is an important cofactor in many metabolic reactions in amino acid metabolism. Those with this deficiency may produce sufficient levels of PAH, but since tetrahydrobiopterin is a cofactor for PAH activity, deficient dihydrobiopterin reductase renders any PAH enzyme non-functional. Tetrahydrobiopterin is also a cofactor in the production of L-DOPA from tyrosine and 5-hydroxy-l-tryptophan from tryptophan, which must also be supplemented as treatment in addition to the supplements for classical PKU. Levels of dopamine can be used to distinguish between these two types. Low levels of dopamine lead to high levels of prolactin. By contrast, in classic PKU, prolactin levels would be relatively normal. Tetrahydrobiopterin deficiency can be caused by defects in four different genes.
The mean incidence of PKU varies widely in different human populations. Caucasians are affected at a rate of 1 in 10,000. Turkey has the highest documented rate in the world, with 1 in 2,600 births, while countries such as Finland and Japan have extremely low rates with fewer than one case of PKU in 100,000 births.