Sex Chromosome Nondisjunction in Humans
Humans display dramatic deleterious effects with autosomal trisomies and monosomies. Therefore, it may seem counterintuitive that human females and males can function normally, despite carrying different numbers of the X chromosome. Rather than a gain or loss of autosomes, variations in the number of X chromosomes are associated with relatively mild effects. In part, this occurs because of a molecular process called X inactivation. Early in development, when female mammalian embryos consist of just a few thousand cells (relative to trillions in the newborn), one X chromosome in each cell inactivates by tightly condensing into a quiescent (dormant) structure called a Barr body. The chance that an X chromosome (maternally or paternally derived) is inactivated in each cell is random, but once the inactivation occurs, all cells derived from that single cell will have the same inactive X chromosome or Barr body.
By this process, a phenomenon called dosage compensation is achieved. Females possess two X chromosomes, while males have only one; therefore, if both X chromosomes remained active in the female, they would produce twice as much product from the genes on the X chromosomes as males.
So how does X-inactivation help alleviate the effects of extra X chromosomes? An individual carrying an abnormal number of X chromosomes will inactivate all but one X chromosome in each of her cells. If three X chromosomes are present, the cell will inactivate two of them. If four X chromosomes are present, three will be inactivated, and so on. This results in an individual that is relatively phenotypically normal. However, even inactivated X chromosomes continue to express a few genes, and X chromosomes must reactivate for the proper maturation of female ovaries. As a result, X-chromosomal abnormalities are typically associated with mild mental and physical defects, as well as sterility. If the X chromosome is absent altogether, the individual will not develop in utero.
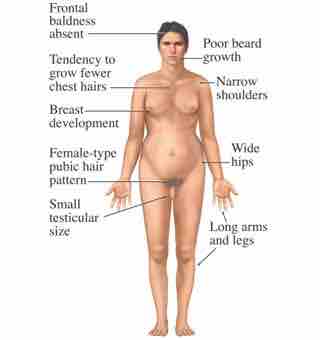
Several errors in sex chromosome number have been characterized. Individuals with three X chromosomes, called triplo-X, are phenotypically female, but express developmental delays and reduced fertility. The XXY genotype, corresponding to one type of Klinefelter syndrome, corresponds to phenotypically male individuals with small testes, enlarged breasts, and reduced body hair . More complex types of Klinefelter syndrome exist in which the individual has as many as five X chromosomes. In all types, every X chromosome except one undergoes inactivation to compensate for the excess genetic dosage. This can be seen as several Barr bodies in each cell nucleus. Turner syndrome, characterized as an X0 genotype (i.e., only a single sex chromosome), corresponds to a phenotypically female individual with short stature, webbed skin in the neck region, hearing and cardiac impairments, and sterility.
Sex Chromosome Nondisjunction
The symptoms of Klinefelter's syndrome (XXY) in a human male.
Duplications and Deletions
In addition to the loss or gain of an entire chromosome, a chromosomal segment may be duplicated or lost. Duplications and deletions often produce offspring that survive but exhibit physical and mental abnormalities. Duplicated chromosomal segments may fuse to existing chromosomes or may be free in the nucleus. Cri-du-chat (from the French for "cry of the cat") is a syndrome associated with nervous system abnormalities and identifiable physical features that result from a deletion of most of 5p (the small arm of chromosome 5) . Infants with this genotype emit a characteristic high-pitched cry on which the disorder's name is based.
Cri-du-chat Syndrome
This individual with cri-du-chat syndrome is shown at two, four, nine, and 12 years of age.